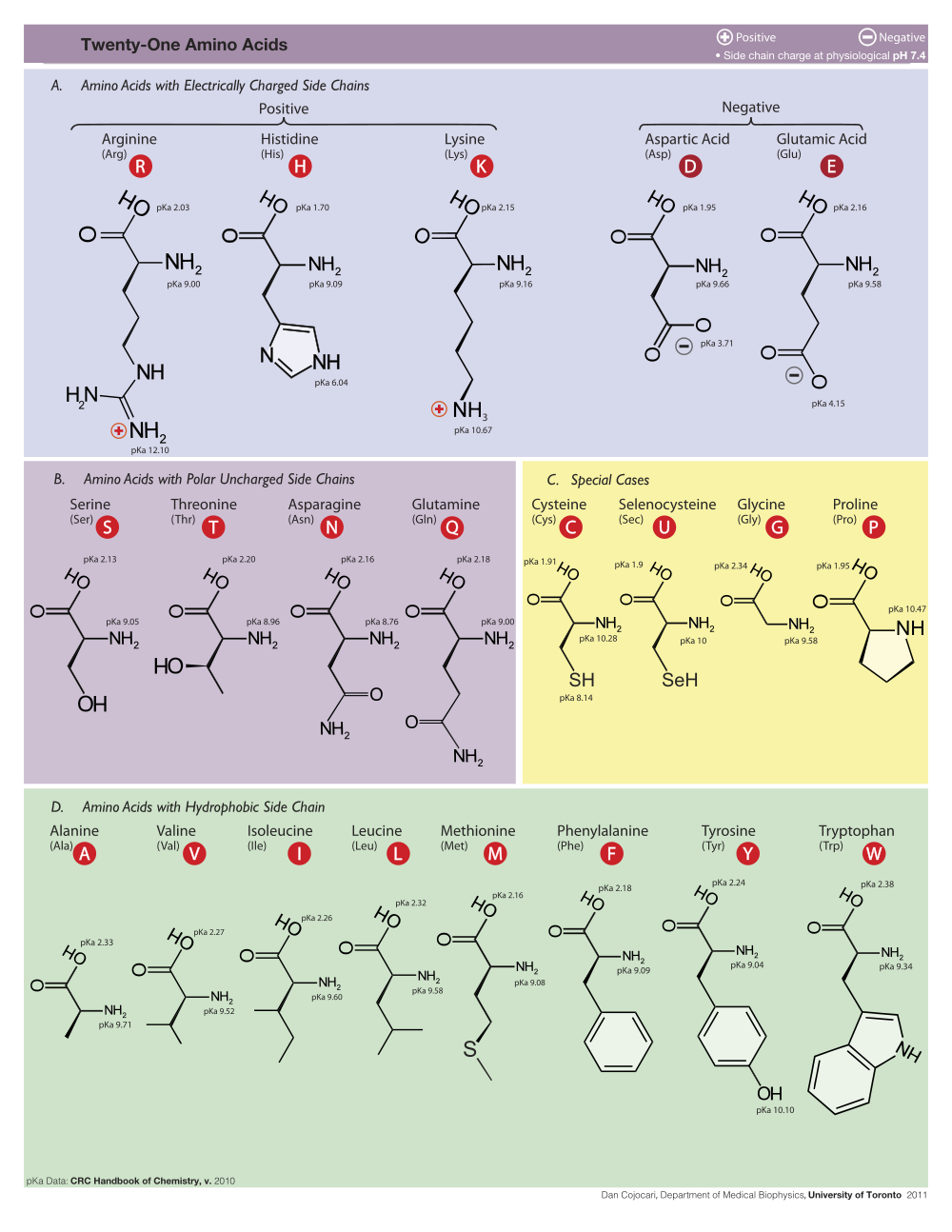
 Clicking on an atom produces an identification
report in the Jmol rectangle, to the lower left of the molecule (see
snapshot at right), and also in the browser's status bar.
Amino acids are reported as
3-letter abbreviations,
and nucleotides as
Clicking on an atom produces an identification
report in the Jmol rectangle, to the lower left of the molecule (see
snapshot at right), and also in the browser's status bar.
Amino acids are reported as
3-letter abbreviations,
and nucleotides as
A, DA, C, DC, G, DG, T, DT, U, or DU (D for Deoxy).
Ligands and non-standard residues have other abbreviations.
Atoms are identified using PDB file format nomenclature, as follows:
|
CA = Carbon, Alpha (1st) CB = Carbon, Beta (2nd) CG, OG = Carbon/Oxygen, Gamma (3rd) CD = Carbon, Delta (4th) CE, NE, OE = Carbon/Nitrogen/Oxygen, Epsilon (5th) NZ = Nitrogen, Zeta (6th) NH = Nitrogen, Eta (7th) |
O3', O5' = 3' and 5' pentose oxygens C1' to C5' = 1' to 5' pentose carbons C2-C8, N1-N9, O2-O6 = elements in bases C5M = C5 Methyl (in Thymine) |
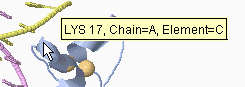 In addition to clicking, you can simply touch an atom with the mouse
(without moving the mouse, which is called "hovering"), and after a
delay of about two seconds, a yellow message will appear near the
mouse pointer (see snapshot at right) with an atom identification
report. This works best with Spin off.
In addition to clicking, you can simply touch an atom with the mouse
(without moving the mouse, which is called "hovering"), and after a
delay of about two seconds, a yellow message will appear near the
mouse pointer (see snapshot at right) with an atom identification
report. This works best with Spin off.
 These labels or marks are shown by default, but can be hidden by
unchecking the checkbox Labels: Show in the Focus Box at the bottom of any
of the upper left panels. (You may have to scroll the upper left panel
down to see this checkbox.)
These labels or marks are shown by default, but can be hidden by
unchecking the checkbox Labels: Show in the Focus Box at the bottom of any
of the upper left panels. (You may have to scroll the upper left panel
down to see this checkbox.)










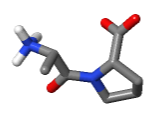



 Water bridge in 1D66 between the sidechain
nitrogen of Arg 46
(chain B)
and a phosphate
oxygen of Arg 46
in Cytosine 13 (chain D).
Water bridge in 1D66 between the sidechain
nitrogen of Arg 46
(chain B)
and a phosphate
oxygen of Arg 46
in Cytosine 13 (chain D).
 appears below the molecule when any of the Unusual components or features
listed below are present.
appears below the molecule when any of the Unusual components or features
listed below are present.
 Problem. FirstGlance will often fail to display
anomalous atoms appropriately. Some anomalous atoms could be invisible
in all views except Vines (with details on).
Problem. FirstGlance will often fail to display
anomalous atoms appropriately. Some anomalous atoms could be invisible
in all views except Vines (with details on).